![]()
![]()
![]()
Глава 1. ОБЗОР ЛИТЕРАТУРЫ.
1. Гены супрессоры опухолевого роста.
1.1 Механизмы действия генов супрессоров.
1.2 Пути повреждения генов супрессоров.
1.2.1 Потеря гетерозиготности.
1.2.3 Метилирование, как механизм инактивации генов супрессоров.
1. ГЕНЫ-СУПРЕССОРЫ ОПУХОЛЕВОГО РОСТА.
В настоящее время представления о генетической природе развития онкологических заболеваний основаны на предположении о существовании генов, нормальная функция которых связана с подавлением опухолевого роста. Такие гены были названы генами-супрессорами опухолевого роста (Weinbrg et al., 1991). Дефекты этих генов приводят к прогрессии, а восстановление функции – к существенному замедлению пролиферации или даже реверсии развития опухоли. Примерами генов-супрессоров служат: ген ответственный за развитие ретинобластомы - ген Rb1, два гена, отвечающие за развитие рака молочной железы - гены BRCA2 и BRCA1, также к генам-супрессорам можно отнести ген WT1 – повреждения которого приводят к нефробластоме, гены CDKN2A и CDKN2B ответственные за развитие меланомы и гематологических опухолей, и другие гены. Инактивация гена hMLH1 приводит к возникновению карциномы желудка и толстого кишечника.
Согласно 2-х ударной модели канцерогенеза, предложенной Knudson в 1971 году, для перехода нормальной клетки в опухолевую необходимо два мутационных события (“удара”). Первым событием является мутация, приводящая к образованию клетки, для которой повышен риск возникновения опухоли. Такие мутации могут возникать как в соматических, так и в половых клетках. Вторым событием, происходящем в соматической клетке, является мутация в неповрежденном аллеле гена и превращение клетки с повышенным риском трансформации в опухолевую, которая начинает безостановочно делиться, что приводит к возникновению злокачественного новообразования. В дальнейшем гипотеза была подтверждена исследованиями, которые показали, что в ряде случаев в опухолях наблюдается гомозиготность по некоторым маркерам, в то время как в крови по этим же маркерам отмечалась гетерозиготность. Таким образом, было установлено, что потеря неповрежденной копии гена - это второе событие по теории Knudson.
Гены-супрессоры опухолевого роста можно подразделить на две большие группы по занимаемому ими месту в системе функционирования эукариотической клетки. В рамках предложенной Кинзлером и Фогельштайном концепции гены-супрессоры предложено разделять на две группы: гены первой группы получили название "хранителей клеточного цикла" (gatekeepers), а гены второй - "общего контроля" (
caretakers) (Баранова, Янковский 1998). Гены - "хранители клеточного цикла" напрямую вовлечены в его регуляцию. Их белковые продукты способны сдерживать опухолевую прогрессию, ингибируя процессы, связанные с делением клетки. Дефекты "генов общего контроля" приводят к повышению нестабильности генома, увеличению частоты возникновения мутаций, и, следовательно, к повышению вероятности повреждения генов, в том числе и "хранителей клеточного цикла".К группе “хранителей клеточного цикла” (ХКЦ) относят такие гены как RB1 (ретинобластома), WT1 (опухоль Вильмса), NF1 (нейрофиброматоз типа I), а также гены, способствующие образованию клеточных контактов, и другие. Если унаследована поврежденная копия гена ХКЦ, образование опухоли может быть инициировано соматической
мутацией в неповрежденном аллеле. Поэтому в случае наследственных форм опухолей, когда имеется герминальная мутация, для начала заболевания необходимо всего одно соматическое мутационное событие - повреждение единственного функционального аллеля. Спорадические случаи возникновения опухоли того же типа требуют двух независимых мутационных событий в обоих аллелях. В итоге, для носителей мутантного аллеля вероятность развития данного типа опухоли значительно выше, чем в среднем по популяции.Инактивация генов “общего контроля” (ОК) приводит к дестабилизации генома - повышается вероятность мутации генов ХКЦ. Дефект последних приводит к появлению опухоли. На фоне поврежденного гена ОК продолжается накопление мутаций, инактивирующих другие супрессоры первой или второй группы, что приводит к быстрому росту опухоли.
При семейных случаях развития некоторых видов рака, мутация в одном из аллелей соответствующего гена ОК может быть унаследована от родителей. Для инициации опухолевого процесса требуется соматическая мутация второго аллеля, а также инактивация обоих аллелей какого-либо гена ХКЦ. Таким образом, для развития опухоли в семейном случае необходимы три независимых мутационных события. Поэтому риск развития опухоли для носителей наследственной мутации гена ОК на
порядок меньше, чем риск для носителя поврежденного аллеля гена ХКЦ. Спорадические опухоли обусловлены соматическими мутациями генов ОК. Они встречаются редко и для их возникновения и развития необходимо четыре независимых мутации.Примерами генов ОК служат гены, ответственные за развитие наследуемого неполипозного рака кишечника - гены MSH-2, MLH-1. Также к этой группе можно отнести широкоизвестный ген-супрессор - р53, мутации или делеции которого наблюдаются примерно в 50% всех злокачественных заболеваний
.1.1. Механизмы действия генов-супрессоров.
Ген Rb1 расположен в проксимальном отделе длинного плеча хромосомы 13q14.1, и, занимает 180 т.п.н. геномной ДНК (Wiggs et al. 1988). Он включает промоторную область около 1,5 т.п.н. (Gill et al. 1994) и состоит из 27 экзонов. Ген RB1 кодирует мРНК длиной 4,7 тысяч нуклеотидов (Mc Gee et al. 1989), экспрессирующуюся в норме во всех клетках организма.
рRb - ядерный фосфопротеин, является негативным регулятором клеточной пролиферации. Активация белка зависит от степени его фосфорилирования и носит в клетках циклический характер (Buchkovich et al. 1989; Chen and De Caprio 1989). В нормальных клетках pRb экспрессируется на протяжении всего клеточного цикла, но белок в различной степени подвергается посттрансляционной модификации, которая регулируется путем фосфорилирования и дефосфорилирования с помощью циклин-зависимых РБ-киназ и фосфатаз. Слабое фосфорилирование происходит в GO и в G1 фазах клеточного цикла, тогда как полное фосфорилирование белка осуществляется киназами на границе перехода G1 в S стадию клеточного цикла. Белок рRb активен только в слабофосфорилированном состоянии (Ladlow et al. 1989; Buchovich et al. 1989; De Caprio et al. 1989). В нефосфорилированном или гиперфосфорилированном состоянии белок рRb не активен, т.е. не может связяться с фактором транскрипции E2F. Анализ фосфорилирования pRb in vitro и in vivo показал, что фосфорилирование pRb в ходе клеточного цикла способны осуществлять активированные протеинкиназы р33cdk2, p34cdk4 и p34cdk2.
Продукт гена Rb1 регулирует экспрессию различных клеточных генов, контролирующих рост клеток (рис. 1). В активном гипофосфорилированном состоянии белок Rb способен связывать фактор инициации транскрипции E2F (Dyson 1998), который является регулятором транскрипции большого числа клеточных генов (например: ДНК полимераза II, DHFR – дигидрофолатредуктаза, р19), ответственных за пролиферацию. В комплексе с pRb этот фактор не способен связываться с промоторной областью генов в результате чего происходит ингибирование транскрипции и задержка клетки в G
1 фазе клеточного цикла (Сhellapan et al. 1991). Фосфорилирование pRb на этом этапе осуществляется циклин-зависимыми киназами cdk4 и cdk6, связанными с D-циклином. Белок р16/Ink4A ингибирует связывание циклинов D-типа с киназами cdk4/6, контролируя, таким образом, переход клетки из фазы G1 в фазу S.Через различные промоторные элементы, такие как семейство Sp транскрипционных факторов, pRb регулирует общую экспрессию ядерных белков (Kennett et al. 1997). Но, в тоже время, белок pRb увеличивает активность некоторых факторов транскрипции (например, C/EBP), участвующих в конечных этапах клеточной дифференцировки (Lee and Lee 1997). Таким образом, pRb выполняет супрессорную роль в опухолеобразовании - препятствует пролиферации, способствует окончательной дифференцировке клеток.
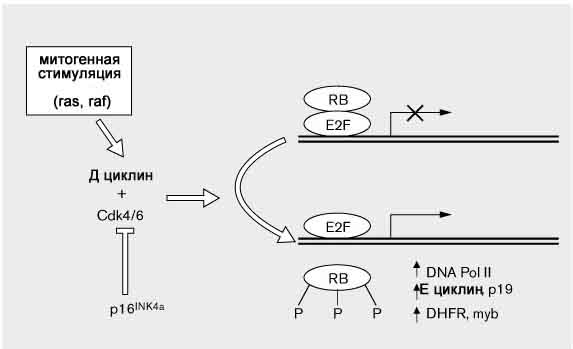
Рисунок 1. Схема действия ретинобластомного белка (pRb)
В вирустрансформированных клетках C-концевая область pRb способна формировать специфические комплексы с трансформирующими белками некоторых ДНК-содержащих онкогенных вирусов, включая большой Т клеточный антиген вируса SV40, белки аденовируса E1A и человеческого вируса папилломы E7 (De Caprio et al. 1988; Whyte et al. 1988; Dyson et al. 1989; Munger et al. 1989). Эти белки являются факторами, активирующими промоторы генов-регуляторов клеточной пролиферации: c-myc, N-myc, ELF1, PU1, c-myb и ряда других генов. Экспрессия протоонкогенов в нормальной клетке необходима для инициирования клеточной пролиферации и начало их работы приходится как раз на Gо® G1 стадии цикла. Продукт гена Rb1 является негативным регулятором экспрессии этих генов (Pientenpol et al. 1990; Robbins et al. 1990).
В последнее время большое значение придается эпигенетической регуляции генной экспрессии. Некоторые продукты генов-супрессоров опухолевого роста влияют на хроматиновую структуру так, что это может приводить к активации или репрессии транскрипции. Структура хроматина может изменяться в результате посттрансляционной модификации ее компонентов. Например, ацетилирование и деацетилирование лизиноых остатков N-концевых участков гистонов.
В 1999 опубликован ряд работ (Struhl et al., Bremh and Kouzaridies) в которых показано участие белка RB в деацетилировании белков. Белок RB может связываться с двумя белками HDAC (histone deacetilase) семейства – HDAC1 и HDAC2. Luo (1999) используя метод иммунопреципитации (CHIP) показали, что статус ацетилирования гистонов изменяется после связывания с pRB. В эксперименте in vitro показано, что во время G0 и G1 фазы клеточного цикла формируется тройной комплекс состоящий из фактора транскрипции E2F, белка RB, активированной деацетилазы (HDAC1), а также других транскрипционных факторов (рис. 2). HDAC1 воздействует на нуклеосомы окружающие промоторные области генов, активных в S фазу, и препятствует их ацетилированию. Это влияние индуцирует изменение конформационной структуры компонентов хроматина, и формируется, так называемая, “закрытая” форма хроматина, что вызывает инактивацию транскрипции.
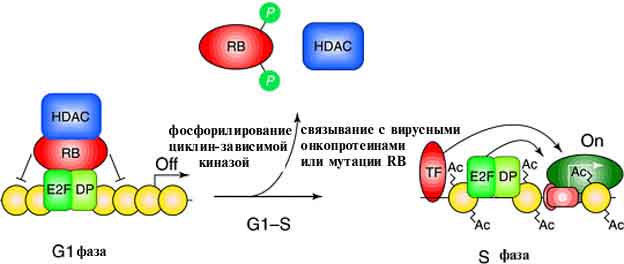
Рисунок 2. Схема участия белка RB в деацетилировании белков.
При переходе клетки из фазы G1 в фазу S, происходит гиперфосфорилирование белка RB, вследствие чего комплекс E2F-pRB-HDAC1 распадается. Это приводит к ацетилированию гистонов и формированию “открытой” – активной структуры хроматина. Транскрипционные факторы (ТФ) получают доступ к промоторной области генов, и происходит активация транскрипции.
Таким образом, степень фосфорилирования pRb определяет механизм контроля инициации транскрипции генов, ответственных за клеточную пролиферацию. В случае отсутствия данного белка или невозможности осуществления его функции происходит сбой в регуляции деления клетки, что приводит к опухолевой трансформации.
Короткое плечо 9 хромосомы часто повреждается при многих видах раковых образований. В локусе INK4A/ARF (9p21) клонировано и картировано два гена: р16/INK4A/CDKN2A/MTS1 и р19/ARF.
Сначала был идентифицирован ген р16, как ингибитор циклин-зависимых киназ, точковые мутации которого были определены в случае семейной меланомы и также показаны гомозиготные делеции в разнообразных опухолевых клетках. Позже в этом же локусе был идентифицирован и второй транскрипт – р19/ARF (ARF – alternate reading
frame – альтернативная рамка считывания). Этот ген кодирует регулятор клеточного цикла, предотвращающий деградацию белка р53.Эти два транскрипта кодируются различными первыми экзонами (1 экзон гена р19 лежит ~13kb центромернее 1-го экзона р16), но одинаковыми 2 и 3 экзонами. Однако белки р16/INK4A и p19/ARF имеют разные рамки считывания в экзоне 2 и, поэтому, между ними не имеется никакого аминокислотного тождества.
Ген CDKN2A/р16/Ink4A/MTS1 является геном-супрессором опухолевого роста и относится к группе “хранителей клеточного цикла”. Показано, что ген р16 играет важную роль в процессе канцерогенеза. Мыши, у которых отсутствует экспрессия гена р16, в раннем возрасте часто развиваются фибросаркомы и лимфомы, а также они имеют повышенную чувствительность к канцерогенам (Serrano et al. 1996). Отмечено участие этого гена в развитии как спорадичаской, так и семейной формы меланомы (Coleman et al. 1994), глиомы (Olopade et al. 1992), при раке легких и Т- и В-клеточных лейкозах (Lekeis et al. 1990). Найдены делеции этого гена (Kamb 1995) в 133 из 260 клеточных линий различных тканевых типов (астрацитома, глиома, лейкоз, меланома, остеосаркома, рак мочевого пузыря, легких, молочной железы, яичника, почки), что составляет от 25 до 82% исследованных линий соответствующего типа. Исключение составили только 20 нейробластомных линий и 10 линий из клеток рака кишечника – ни в одной из них делеций в локусе 9р21 не обнаружено.
В 14 из 34 меланомных клеточных линий, несущих по одной копии данного гена, обнаружены точковые мутации второго аллеля. Почти треть соматических мутаций гена представлена делециями, в то время как среди герминальных мутаций делеции и инсерции составляют не более 5% (Herman et al. 1995, Gonzzalez-Zulueta et al. 1995). Были определены точковые мутации гена р16 при меланоме: Ile41Thr, Arg50Ter, Asn63Ser, Arg79Pro, Gly93Trp, Val118Asp, Ala140Thr, а также мутация донорного сайта сплайсинга IVS2+1.
На молекулярном уровне действие белка р16 основано на ингибировании регуляторов клеточного цикла. Белок р16 – ингибитор циклин-зависимых киназ и является участником биохимического пути Rb/cyclinD/cdk4/p16
INK4a (Dyson and Balmain 1999). р16 связывается с киназами cdk4/6, и этот комплекс нарушает взаимодействие киназ с циклином Д (Rosso et al. 1998). Ингибирование функций циклин-зависимых киназ приводит к гипофосфорилированию белка RB, что уменьшает экспрессию E2F-зависимых генов, блокируется переход клетки из фазы G1 в фазу S (Koh et al. 1995, Serrano et al. 1996, Lukas et al 1995) и осуществляется контроль клеточного деления и пролиферации (рис. 1).Экспериментально показано (Kiyono 1998), что снижение экспрессии р16 приводит к гиперфосфорилированию pRB. В таком состоянии этот белок не может оставаться связанным с фактором транскрипции E2F. Поэтому комплекс RB-E2F диссоциирует и свободный фактор E2F активирует транскрипцию генов, специфичных для S фазы (S-phase-specific-gene).
Белок р19
ARF у человека состоит из 132 аминокислот и имеет молекулярную массу 14 КДа. Этот же белок у мышей состоит из 169 аминокислот и не имеется никакой гомологии с другими мышиными белками. Молекулярная масса этого белка составляет 19 КДа.Функция р19 состоит в том, что в активированном состоянии этот белок ингибирует фактор MDM2, который является фактором связывания белка р53 с убиквитином, что предотвращает его деградацию. Активация р19 происходит в ответ на действие таких онкогенных стимулов как E1A, ras, myc, V-abl, E2F-1. Онкогенный белок
ras является одной из причин преждевременного старения фибробластов эмбрионов мышей, а myc и E1A – стимулируют апоптоз. Показано, что эти онкогены обеспечивают активацию p19ARF/p53/MDM2 пути, который запускается при иммортализации клеток грызунов in vitro (de Stanchina et al. 1998, Zindy et al. 1998, Palmero et al. 1998).Основными соматическими мутациями, повреждающими р19/ARF являются делеции. Гомозиготные делеции этого гена обнаруживаются в 19% случаев опухолей, в то время как точковые мутации составляют не более 3% (Sharpless et DePinho 1999). Так как гены р16/INK4A и p19/ARF кодируются одной последовательностью ДНК, очень часто инактивируются оба гена вследствие одного и того же повреждения.
Гены BRCA1 и 2 являются также генами-супрессорами опухолевого роста и относятся к группе "генов общего контроля". Эти гены отвечают за развитие некоторых форм рака молочной железы. Мутации одного из этих генов (BRCA1) наблюдаются в 5-10% всех опухолей данного типа, однако, в их фенотипическом проявлении имеются некоторые различия. Так, мутации BRCA2 (но не BRCA1) связаны с повышенным риском развития рака молочной железы у мужчин, а для носителей аберрантного аллеля BRCA1 значительно повышена вероятность развития рака яичников и в меньшей степени раков простаты и кишечника (Ford et al. 1994). Продукты обоих генов способны взаимодействовать с белком RAD51 (Scully et al. 1997, Sharan et al. 1997), являющимся гомологом бактериального белка RecA, основного белка системы репарации двухцепочечных разрывов ДНК. Для продуктов BRCA1 и RAD51 показана совместная локализация в составе синаптонемальных комплексов мейотических хромосом. Эмбриональные и трофобластные клетки, дефектные по экспрессии BRCA2, сверхчувствительны к ионизирующей радиации. Эти данные говорят о том, что нарушения репарационного пути BRCA/RAD51 могут вести к увеличению нестабильности генома
.Оба гена играют важную роль в процессах эмбрионального развития. Мыши, у которых инактивированы обе копии гена – гомолога BRCA1 либо BRCA2, гибнут на ранних стадиях внутриутробного развития, при этом носители гемизиготной мутации развиваются нормально (Scully еt al., Sharan et al 1997). Таким образом, отсутствие экспрессии этих генов-супрессоров ведет скорее к клеточной смерти, чем усиленной пролиферации клеток. Очевидно, существуют механизмы, непосредственно перед клеточным делением отслеживающие, прошла ли репарация имеющихся повреждений ДНК, и в случае нарушения системы репарации, останавливающие деление или даже направляющие клетку к гибели путем апоптоза (Marx еt al. 1997).
Не исключено, что мутации генов BRCA1 и BRCA2 могут приводить к образованию опухолей не только описанным выше путем. Оба белка достаточно велики – 1863 аминокислотных остатков в продукте гена BRCA1 и 3418 – в BRCA2 и могут содержать домены, нужные для выполнения других функций, не связанных с репарацией ДНК. Так, найдена гомология
между третьим экзоном гена BRCA2 и активационным доменом c-Jun, содержащим сайт для JNK-киназы (Milner et al. 1997). Способность этого домена белка BRCA1 к активации транскрипции подтверждена экспериментально (Milner et al. 1997). Не менее важно, что происходящая в пределах третьего экзона замена Tyr на Cys, приводящая к нарушению способности активировать транскрипцию, наблюдалась в нескольких семейных случаях развития рака молочной железы. По соседству с активационным доменом в белке BRCA2 расположены два домена, способных ингибировать активаторную функцию этого белка. Подобный тип регуляции показан для нескольких транскрипционных факторов, в том числе для белка c-Fos. Возможно, что в случае этого белка его транскрипционная активность запускается после его стимуляции с помощью JNK-подобной киназы. Домен, способный к активации транскрипции, содержится и в составе BRCA1 (Chapman, Varo et al. 1996). Обнаружено также связывание белка BRCA1 in vivo с ранее неизвестным белком BARD1, содержащим, как и BRCA1, обогащенный цистеином RING-домен. Миссенс-мутации, выявленные в некоторых семейных случаях развития рака молочной железы, нарушают это взаимодействие, что говорит о том, что белок BARD1 также может быть вовлечен в опухолевый процесс (Wu et al. 1996).К группе “генов общего контроля” относят и те гены-супрессоры, продукты которых не входят в системы репарации ДНК непосредственно, а принимают участие в организации “контрольного пункта” проверки ДНК перед переходом клетки к следующей стадии клеточного цикла, обеспечивающего две основных контролирующих функции: 1) проверку того, что предыдущая стадия завершена полностью, и 2) в случае необходимости создания возможности для прохождения репарации ДНК перед началом репликации (Hartwell et al. 1994). В частности, это означает, что разрывы в цепи ДНК приводят к задержке клеточного цикла в фазе G1, препятствуя переходу клетки в фазу S (Nelson et al. 1994). Ключевую роль в этом процессе играет один известный ген-супрессор – p53, мутации или делеции которого наблюдали примерно в 50% всех злокачественных заболеваний (Greenblatt et al. 1994). Ген р53 картирован и клонирован на коротком плече хромосомы 17р13 и занимает 200 т.п.н. геномной ДНК, состоит из 11 экзонов, первый из которых является некодирующим. Ген кодирует мРНК длиной 2,8 тысяч нуклеотидов. Белок массой 53Кда является фосфопротеином и состоит из 393 аминокислот.
Белок р53, функционирующий в виде тетрамера, связывается с убиквитином (Ub) при участии фактора MDM2 (murine double minute 2) и подвергается деградации (рис. 3). Белок р19/ARF, экспрессия которого увеличивается под действием онкостимулами типа E1a, V-abl, myc, нарушает это связывание и предотвращает деградацию р53. Также ингибитором этого связывания является продукт гена АТМ, повреждения которого
вызывают атаксию-телангиоэктазию.Белок p53 способен функционировать также как активатор транскрипции. Показано, что множественные повреждения ДНК вызывают экспрессию гена p53 (Nelson et al. 1994), который, в свою очередь, активирует транскрипцию гена р21/WAF1, кодирующего ингибитор циклин-зависимых киназ CIP1 (Dulic et al 1995, El-Deiry et al. 1994). CIP1 избирательно подавляет активность циклин D1/CDk4 и циклин E/CDk2-комплексов, что и приводит к задержке перехода клетки из фазы G1 в фазу S.
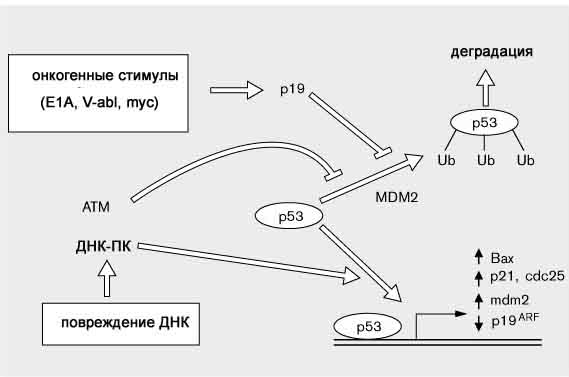
Рисунок 3. Схема действия белка р53.
Запускающая этот каскад индукция p53 происходит в ответ на любые неспецифические, повреждающие ДНК, воздействия, такие как ионизирующая радиация, свободные радикалы, действие эндонуклеаз рестрикции (Nelson et al. 1994). Одновременно с подавлением репликации ДНК, белок p53 стимулирует ее репарацию, активирует транскрипцию гена GADD45 (Smith et al. 1994), а также связывается с белком ERCC3, непосредственно участвующем в распознавании и вырезании поврежденных участков ДНК (Wang et al. 1994).
Герминальные миссенс-мутации гена-супрессора p53 приводят к довольно редкому синдрому Ли-Фраумени, при котором типичными являются опухоли мозга, мягкотканные саркомы различной локализации, остеосаркомы, лейкемия и карциномы молочной железы. Выявлена кластеризация мутаций в районе 14 кодонов (245-258) (Vogelstein 1990). В то же время различные повреждения гена выявляют практически во всех типах опухолей (Levine 1991). Частота миссенс-мутаций гена составляет 74%, сдвига рамки считывания - 11%, нонсенс-мутаций - 7%, мутаций сайта сплайсинга - 4%. Определено 7 “горячих” точек, повреждаемых мутациями: кодоны 130-142, 151-164, 171-181, 193-200, 213-223, 234-258 и 270-286, причем все они локализованы в эволюционно консервативном ДНК-связывающем домене, кодируемом экзонами 5-8. Мутации кодонов 157 и 179 наиболее часто выявляются при раке легкого, кодона 175 - при раке толстого кишечника, кодона 248 - при сквамозных карциномах головы и шеи, кодона 249 - при гепатокарциноме и кодона 278 - при опухолях кожи (Hussain, 1998).
Повышенный интерес к генам-супрессорам опухолевого роста стимулировал разработку методов их направленного поиска. Ген ING1, кодирующий ядерный белок с молекулярной массой 33 кД (Garkavtsev et al., 1996), был одним из первых генов, обнаруженных методом вычитательной гибридизации с последующим отбором и клонированием фрагментов с трансформирующей активностью. Выборочный анализ клеточных линий показал, что 3’-участок гена ING1 перестроен или содержит делеции в нейробластомной линии SK-N-SH, а содержание белка p33
ING1 снижено в культурах, полученных из злокачественных клеток от больных раком молочной железы (Garkavtsev et al., 1996). Эти данные подтверждали предполагаемую принадлежность гена к супрессорам опухолевого роста.За первой статьей, посвященной гену ING1, последовал целый ряд работ, уточнявших роль белкового продукта гена в клетке и механизмы его действия. Было показано, что ING1, как и некоторые другие гены-супрессоры, участвует в регуляции апоптоза - программируемой клеточной гибели (Helbing 1997). Так, увеличение уровня экспрессии р33/ING1 в полученной из клеток тератокарциномы линии р19, наблюдается при сывороточном голодании, индуцирующем апоптоз. Гиперэкспрессия р33/ING
1 в клеточной линии р19 и фибробластах грызунов, содержащих тетрациклин-зависимый ген с-myc, заметно усиливает Myc-зависимый апоптоз. Напротив, конститутивная экспрессия антисмысловой мРНК гена ING1 препятствует апоптозу в этих клетках. Таким образом, потеря геном ING1 своей функции приводит к клеточной пролиферации и развитию опухоли вследствие сниженной чувствительности к сигналам апоптоза.Точно установлено, что ген ING1 вовлечен в процессы клеточного старения и регуляцию клеточного цикла (Garkavtsev and Riabovol 1997). Содержание белка р33/ING1 и соответствующей ему мРНК существенно повышено в стареющих диплоидных фибробластах человека (в 8-10 раз по сравнению с молодыми), а ингибирование р33/ING1 введением антисмысловой мРНК воспроизводимо приводит к увеличению продолжительности жизни диплоидных фибробластов человека примерно в семь раз. Экспрессия р33/ING1 регулируется на протяжении клеточного цикла и достигает максимума во время синтеза ДНК. Разными методами было показано, что гиперэкспрессия этого белка эффективно блокирует клеточный цикл при переходе из фазы G0 в G1.
Сходство функций продуктов генов ING1 и р53 - гена-супрессора опухолевого роста, определяющего клеточный ответ на разные типы стресса (Gottlieb and Oren 1996) - заставило предположить их принадлежность одному сигнальному пути, что и было подтверждено в работе И. Гаркавцева с сотрудниками (Garkavtsev et al., 1998). Как выяснилось, биологические эффекты этих белков скоррелированы и требуют активности обоих генов: ни один из них не способен
подавлять клеточный рост в одиночку, при инактивации другого. Это объясняется экспериментально обнаруженной зависимостью активации транскрипции с промотора p21/WAF1, ключевого механизма р53-зависимого контроля клеточного роста, от экспрессии ING1. Кроме того, при иммунопреципитации белки p33/Ing1 и p53 могут быть осаждены в составе единого комплекса, что говорит об их непосредственном взаимодействии. Как с помощью FISH-гибридизации (Garkavtsev et al., 1997), так и с помощью ПЦР-скрининга панели радиационных гибридов Stanford G3 (Zeremski et al., 1997), ген ING1 локализован в субтеломерном районе длинного плеча 13-й хромосомы (13q34), изменения которого были зарегистрированы при разных видах рака (Stanbridge 1992). Отчасти высокая частота перестроек длинного плеча 13-й хромосомы объясняется расположением на нем двух генов-онкосупрессоров RB1 и BRCA2 (районы 13q14 и 13q12 соответственно) (Hinds and Weinberg 1994; Wooster et al.,1994). Но в ряде случаев, например, в большой группе чешуйчатоклеточных карцином головы и шеи, перестройки не затрагивали генов RB1 и BRCA2, а потеря гетерозиготности наблюдалась в субтеломерном районе 13-й хромосомы, где расположен ген ING1 (Maestro et al. 1996).Вовлеченность гена ING1 в регуляцию процессов апоптоза и клеточного старения, локализация в подверженном перестройкам районе 13-й хромосомы и сведения о снижении экспрессии ING1 и перестройках гена, обнаруженных в некоторых клеточных линиях, полученных из опухолевых клеток (Garkavtsev et al., 1996), привлекли внимание многих исследователей. О перспективности изучения структуры гена и его белкового продукта свидетельствовали также наличие высококонсервативных гомологов ING1 у мыши и дрожжей (Zeremski et al., 1999; Баранова и др, 2000).
За относительно короткое время появились данные по мутационному статусу гена ING1 и по степени его экспрессии в различных типах опухолей. Показано, в частности, что перестройки субтеломерного района длинного плеча 13-й хромосомы, сопровождающие чешуйчатоклеточные карциномы головы и шеи, не затрагивают ING1 (Sanchez-Cespedes et al., 2000). На 452 образцах карцином молочной железы был проведен мутационный и экспрессионный анализ гена ING1 (Jager et al., 1999; Toyama et al. 1999). Установлено, что мутирует он очень редко, а вот заметное (в 2 - 10 раз по
сравнению с нормальной тканью) снижение его экспрессии наблюдалось в 44% опухолей и в 10 из 10 исследованных клеточных линий. Более того, большинство опухолей (58%) со сниженной экспрессией ING1 давали метастазы в лимфоузлы, в то время как среди опухолей с повышенной экспрессией гена таких было только 9%. Анализ экспрессии гена ING1 при раке желудка выявил ее значительное снижение в 15 из 20 исследованных случаев. Из 12 клеточных линии точковые мутации были обнаружены только в одной (Oki et al., 1999). Аналогичная ситуация наблюдалась и в случае глиом (Shinouraet al., 1999), Т- и В-клеточных лейкозов (Ohmori et al., 1999) и карцином прямой кишки (Sarela et al., 1999): для них показано общее снижение экспрессии гена ING1 при наличие обоих аллелей гена и отсутствии точечных мутаций и делеций. Это предполагает снижение экспрессии гена за счет транскрипционных и посттранскрипционных механизмов.Совокупность этих данных не противоречит возможной роли гена как супрессора опухолевого роста, утрата или инактивация которого приводит к образованию опухоли, однако, до настоящего времени остается неясным, ассоциированы ли мутации гена ING1 с какими-либо конкретными типами опухолей.
В результате автоматизированного определения нуклеотидной последовательности обнаружено, что ген ING1 устроен относительно просто. Структура гена ING1 содержит по крайней мере два экзона: 5’-концевой экзон 1a и общий для обеих форм мРНК 3’-концевой экзон 2. Размер интрона между ними составляет 3.5 т.п.н. Изоформа мРНК ING1 1b также состоит из двух экзонов: экзона 1b размером 450 н.п. и общим для обоих форм экзона 2, размером 1845 н.п. Для всех экзон-интронных границ выполняется правило GT/AG, донорный и акцепторный сайты сплайсинга почти точно соответствуют опубликованным консенсусным последовательностям (Sharp et al., 1997).
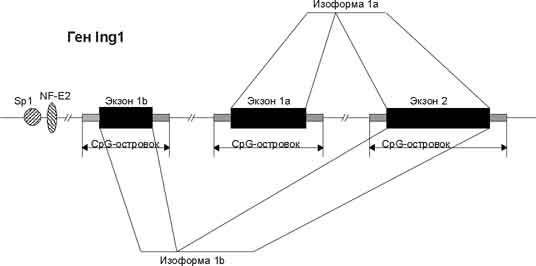
Рисунок 4. Структура гена Ing1.
Анализ нуклеотидной последовательности геномного локуса ING1, в том числе прилегающих к экзонам интронных последовательностей, а также нетранскрибируемых последовательностей, предшествующих экзонам 1a и 1b, показал, что локус ING1 отличается необычно высоким содержанием нуклеотидов G и С. Каждому из трех экзонов ING1 сопутствует выраженный CpG-островок (рис. 4), содержащий множество участков, не распознаваемых метил-чусвтвительными эндонуклеазами рестрикции. GpC-островок экзона 1b размером по крайней мере 933 н.п. содержит 68% C и G, отношение CG к GC составляет 0,99. CpG-островок, включающий экзон 1a и предшествующая ему последовательность содержит 57% C и G, экзон 2 содержит CpG-островок с 59% C и G. Наличие CpG островков может указывать на возможности регуляции экспрессии ING1 путем метилирования ДНК
de novo. Выше предполагаемых начальных позиций экзонов 1a и 1b выявлены элементы Sp1 и NF-E2, связывающие соответствующие белки и защищающие ДНК от метилирования.При В-клеточном хроническом лимфолейкозе (В-ХЛЛ) наиболее частым кариотипическим изменением, обнаруживаемым с помощью стандартных цитогенетичксих методов, являются делеции хромосомы 13 в районе 13q14.3. Следующими по частоте являются структурные изменения в длинных плечах хромосомы 11, 6 и 14, а также трисомия 12.
Работы по поиску гена-супрессора, утрачиваемого или повреждаемого при В-ХЛЛ начались в 1994 г. В 1997 г. были выделены два перекрывающихся клона кДНК (CLL2 и CLL5), представляющих собой кодирующую область нового гена человека, получившего название Leu5. С помощью гибридизации по Саузерну было обнаружено, что кодирующаяя область этого гена расположена на расстоянии
около 60 т.п.н. от границы минимального участка утрачиваемого у больных с В-ХЛЛ. Ген Leu5 кодирует белок в высокой степени гомологичный известным белкам человека, вовлеченным в процессы канцерогенеза, эмбриогенеза и регуляцию иммунного ответа.
При общей длине гена 407 аминокислот в его составе можно выделить три консервативных домена:
Такое сочетание доменов позволяет отнести ген Leu5 к семейтву Rfp – Ret finger protein. Это белковое семейство эволюционно-консервативно, что свидетельствует о его значимости для жизни клетки. Так, близкие гомологи белка Leu5 обнаружены у гребенчатого тритона, шпорцевой лягушки и даже червя Cenorhabditis elegans.
Кроме того, белок Leu5 содержит целый ряд участков подвергающихся посттрансляционным изменениям, к примеру, фосфорилированию протеинкиназой С и другими протеинкиназами, принимающими участие в запуске опухолевого процесса.
В настоящее время удалось клонировать два 5`-концевых нетранслируемых экзона длиной 101 и 65 нуклеотидных пар, отстоящих на значительное расстояние от кодирующей области гена Leu5 – это расстояние составляет не менее 20 т.п.н. Удалось определить хромосомную ориентацию гена – его транскрипция происходит в направлении от центромеры к теломере. Также обнаружено, что между первым и вторым нетранслируемыми экзонами Leu5 расположен выраженный CpG-островок, поэтому возможна регуляция экспрессии этого гена посредством метилирования.
Из всех генов до настоящего времени обнаруженного в исследуемом районе, только клонированный ген Leu5 имеет длинную открытую рамку считывания и значимые гомологии к известным регуляторным белкам человека, позволяющие высказать предположения о его функции. Поэтому ген Leu5 является первостепенным кандидатом на роль супрессора опухолевого роста, вовлеченного в развитие В-ХЛЛ.
1.2. Пути повреждения генов-супрессоров опухолевого роста.
Повреждение генов-супрессоров приводит к возникновению или прогрессирующему росту опухолей. В нестоящее время известно, по крайней мере, 3 основных механизма инактивации генов-супрессоров:
1.2.1. Потеря гетерозиготности.
Потеря гетерозиготности – частное событие, происходящее в опухоли; может быть вызвано делецией участка или полной потерей хромосомы.
Согласно 2-х ударной модели канцерогенеза, предложенной Knudson в 1971 году, предполагается, что для перехода нормальной клетки в опухолевую необходимо два последовательных мутационных события. Первым событием является мутация, приводящая к образованию клетки, которая обладает повышенным риском возникновения опухоли. Такие мутации могут возникать как в соматических, так и в половых клетках. Вторым событием, происходящим в соматической клетке, является мутация в неповрежденном аллеле или потеря гетерозиготности гена и, затем, превращение клетки в злокачественную. Примером могут служить случаи потери гетерозиготности (ПГ) локуса 11р15 при нефробластоме (Reev et al. 1989), а также ПГ локуса 9р21 в случае меланомы (Healy et al 1995), ПГ локуса 1р36 при нейробластоме (Fong et al. 1989) и ПГ локуса 8р21-12 при раке простаты (Trapman et al. 1994). Анализ потери гетерозиготности в опухолях, развивающихся при достаточно редком аутосомно-доминантном заболевании (1 на 100 тыс.) – туберозном склерозе, показал, что теряются маркеры, расположенные в коротком плече хромосомы 16p13.3 (чаще) и в длинном плече хромосомы 9q34 (реже) (Carbonara 1996). Во всех опухолях у пациентов с синдромом Горлина и в спорадических базально-клеточных карциномах обнаруживается ПГ по маркерам района хромосомы 9q22.3 (Levanat 1996). Исследование ПГ гена Rb1 (локус 13q14.1) в ретинобластомах по двум внегенным и одному внутригенному маркерам показало ПГ в 70% случаев (Бабенко 2000).
Потеря гетерозиготности (ПГ) может являться результатом:
Под мутацией понимают все изменения в последовательности ДНК, независимо от их локализации и влияния на жизнеспособность особи.
По типу изменения в нуклеотидной последовательности выделяют мутации сдвига рамки считывания, приводящие либо к образованию бессмысленного белка, либо к преждевременному окончанию его синтеза, и нонсенс-мутации, представляющие собой замены нуклеотидов, при которых образуются терминирующие кодоны, с наибольшим повреждающим действием. Мутации сдвига рамки считывания возникают вследствие делеции или инсерции, некратные трем нуклеотидам. Проявление таких мутаций зависит от их локализации. Чем ближе мутации к началу гена, то есть к началу транскрипции, тем короче их белковые продукты, которые не способны к модификациям и быстро деградируют. 77% всех мутаций гена АТМ и 64% мутаций гена BRCA1 являются мутациями сдвига рамки считывания. Основным типом мутаций выявляемых в гене Rb1 являются нонсенс-мутации, такие же мутации определены в генах р53 (7%) (Hussain and Harris 1998). Семейный аденоматозный полипоз желудочно-кишечного тракта - аутосомно-доминантное заболевание, вызываемое мутациями в гене APC. Вариантом заболевания является синдром Гарднера, при котором преимущественно поражается толстый кишечник. При заболевании достаточно часто выявляются гепатобластомы, опухоли щитовидной железы, медуллобластомы, фибромы и другие опухоли (Gorlin 1960). Основным типом мутаций являются делеции и нонсенс-мутации, приводящие к преждевременной терминации синтеза белка. Герминальные мутации преимущественно повреждают первую половину гена, причем обнаружено два “горячих” кодона - 1061 и 1309. Мутации последнего кодона приводят к раннему появлению полипов и быстрому их озлокачествлению. Соматические мутации в основном локализуются в районе кодонов 1286-1513, с преимущественным повреждением кодонов 1309 и 1450 (Beroud 1996, . Lamlum 1999, Wallis 1999).
Мутации на стыке экзонов и интронов - мутации сайтов сплайсинга - часто нарушают процессинг первичного РНК-транскрипта, в результате чего происходит либо неправильное вырезание соответствующей интронной области и трансляция бессмысленного удлиненного белка, не защищенного от протеолитического действия внутриклеточных ферментов, либо вырезания экзонов и образование делетированного белка. Во всех случаях мутации сайтов сплайсинга, как правило, обусловливают тяжелое течение болезни. Наиболее часто мутации затрагивают существенные инвариантные GT и AG динуклеотиды, локализованные соответственно в начале интрона (донорный сайт сплайсинга) и в конце интрона (акцепторный сайт сплайсинга). В результате чего происходит потеря экзона. Критическое значение имеют также последовательности, прилежащие к указанным динуклеотидам. Так в гене р53 они выявлены всего в 4% случаев, в гене АРС – в 6%, в гене BRCA1 – 2% случаев (Hussain and Harris 1998). Из 16 мутаций, выявленных в гене EXT1, ответственного за возникновение множественной экзостозной хондродисплазии, (Чеснокова 1999) 10 из них относятся к мутациям сдвига рамки считывания и сайтов сплайсинга.
В случае миссенс-мутаций наиболее часто замены, приводящие к изменениям на уровне белка, затрагивают нуклеотидные основания в первой или во второй позиции кодона, однако замены основания в третьей позиции кодона также могут приводить к серьезным нарушениям из-за вероятности активации криптических сайтов сплайсинга. Фенотипическое проявление замен в кодонах миссенс-мутаций зависит от природы соответствующих аминокислотных замен в белке и от функциональной значимости того домена, в котором это произошло. Так, замены аминокислот в активных центрах белков могут сопровождаться полной потерей его функциональной активности, тогда как даже значительно более серьезные нарушения в других частях белка часто оказывают существенно меньшее влияние на фенотип (Горбунова, Баранов, 1997). К высоко мутабильным районам, так называемым “горячим точкам” мутагенеза, относят CpG обогащенные районы и тандемные повторы в кодирующих районах ДНК. Миссенс-мутации в 7-10 экзонах гена WT1, кодирующих мотив цинковых пальцев, определяются у больных с синдромом Дениса-Драша, для которого характерна прогрессирующая нефропатия, псевдогермафродитизм, нефро- и гонадобластомы (Englert 1998). Также показано, что мутации гена р53 в 70% случаев являются миссенс-мутациями. Такие мутации в гене BRCA1 выявлены в 29%, ATM – в 6%, APC – в 16% случаев (Hussain and Harris, 1998). Герминальные миссенс-мутации RET выявлены у пациентов с синдромом МЭН 2A (медуллярный рак щитовидной железы (МРЩЖ), феохромоцитома, гипреплазия паращитовидных желез, ганглионейромы слизистой губ, щек и языка) и семейной форме МРЩЖ, причем мутации в основном затрагивают пять цистеиновых кодонов в 10 и 11 экзонах и на них приходится 97% и 85% всех мутаций, соответственно (Eng 1997). Спорадические мутации в тирозинкиназном домене гена более характерны для синдрома МЭН 2В, отличающегося от типа 2А более агрессивным течением и отсутствием гиперплазии паращитовидных желез и ганглионейром, а точковая мутация в экзоне 16 (T918C) была идентифицирована в 93% случаев (Eng 1997).
Различные изменения в нуклеотидной последовательности транскрибируемых областей ДНК могут по-разному проявляться в фенотипе. Часть из них не оказывает никакого влияния на структуру и функцию соответствующего белка. Примером могут служить замены нуклеотидов, не приводящие к замене аминокислот в силу вырожденности генетического кода. Такие замены принято называть полиморфизмами (или в иностранной литературе – silence mutation). Но не всегда полиморфизмы так “невинны”. Так Siegelmann и Buetow (1998) сообщают о замене гуанина (G) на аденин (A) в кодоне кодирующем валин (Val)80 в гене ароматазы (Cyp 19) кодирующем цитохром Р450. Этот полиморфизм является маркером предрасположенности развития рака груди у женщин. Полиморфизм ДНК в еще одном цитохроме - P450c17
a (CYP17), участвующим в биосинтезе стероидных половых гормонов, так же может повышать риск возникновения рака молочной железы. В 5’ области гена в позиции -27 выявлена однонуклеотидная замена T->C, которая создает сайт узнавания для фермента MspAI и транскрипционного фактора Sp1. Показано, что этот аллель влияет на экспрессию гена и увеличивает риск возникновения рака груди в 2,5 раза (Feigelson 1996, 1998)Большинство мутаций, затрагивающих регуляторные области, выявляют в промоторной части генов, где они изменяют консервативные последовательности. Этот тип мутаций остается пока наименее изученным и, возможно, часть не выявляемых в ряде патологий мутаций относится к этому типу. Мутации, затрагивающие регуляторные области генов сопровождаются, как правило, количественными изменениями соответствующего продукта и не затрагивают структуры и функциональной активности белка. Проявление таких мутаций определяется пороговым уровнем концентрации белка, при котором его функция еще сохраняется. Как правило, регуляторные мутации
менее серьезны и обладают более выраженным плейотропным эффектом по сравнению с мутациями структурных генов.1.2.3. Метилирование, как механизм инактивации генов-супрессоров опухолевого роста.
Еще одним механизмом, приводящим к инактивации генов-супрессоров опухолевого роста, является метилирование промоторной области этих генов. Некоторые гены-супрессоры опухолевого роста содержат в промоторной области CpG-островки. СpG-островки – это CG- и GC-богатые (более 60%) участки ДНК протяженностью от 200 п.н. Примерами таких генов могут служить ген ретинобластомы Rb1, гены CDKN2A и CDKN2B, ответственные за развитие меланомы и гематологических опухолей, соответственно.
В последнее время в 2-х ударную модель канцерогенеза внесены дополнения. Известно, что метилирование промоторной области гена может ингибировать транскрипцию посредством нарушения связывания факторов транскрипции. Кроме того, метилирование CpG-островков может способствовать взаимодействию неспецифических ДНК связывающих белков, таких как MeCP2, способных репрессировать транскрипцию (Nan et al. 1998, Jones et al. 1998).
Суть метилирования заключается в том, что это - химическая модификация, катализируемая ферментом, которая добавляет метильные (CH
3) группы на специфические сайты белков, ДНК и РНК. Реакция ДНК метилирования катализируется ферментом ДНК-метилтрансферазой, который осуществляет перенос метильной группы (CH3) с S-аденозилметионина на цитозин, стоящий перед гуанином. У человека и большинства млекопитающих ДНК метилирование – естественная модификация ДНК, и воздействует только на основание цитозин (С), стоящий перед гуанином (G), т. е. метилирование происходит только в CpG-динуклеотидах. У растений 5-метилцитозин можно обнаружить в динуклеотидах CG и тринуклеотидах CNG (Т-С, А или Т). 70-80% всех CpG-динуклеотидов в человеческом геноме метилированы. Однако в нормальной ткани метилирование происходит, прежде всего, в областях, где плотность CpG динуклеотидов низка, и большинство CpG-островков в норме полностью неметилированы. Но есть исключения из этого правила: метилированные промоторные области генов на инактивированной Х хромосоме, например, ген FMR1 (Li et al 1993, Singer-Sam et al. 1993) и импринтированные гены. Так экспрессия таких генов как IGF2, SNRPN, Znf-21, Par-5 идет только с отцовской хромосомы, а генов WT1, Mash2, p57 – только с материнской (Vogelstein et Kinzler ). Также профиль метилирования может меняться в течение жизни организма. Некоторые гены, экспрессирующиеся в эмбриональном периоде, перестают функционировать к моменту рождения особи. А для таких генов как E-CAD и DBCCR1 показано метилирование в нормальных тканях по мере старения организма.Функция ДНК метилирования до сих пор не полностью ясна. За последние годы было предложено несколько версий роли ДНК метилирования: контроль экспрессии гена, контроль целостности хромосомы, контроль пререкомбинантных событий. Гипотезой о роли ДНК метилирования в геноме является предположение о том, что это защитный механизм против встраивания в геном “паразитных” последовательностей типа ретровирусных элементов (Issa , 1998).
К структурам, обеспечивающим этот процесс, относятся: несколько ДНК-метилтансфераз, деметилазы, центры метилирования, которые инициируют метилирование, и вероятно, связаны с необычными третичными структурами ДНК. Эмбрионы мышей, несущие нонсенс мутацию в гене метилтрансферазы, не развиваются дальше 8-го сомита, но при этом наблюдается низкий, но постоянный уровень метилцитозина (Lei et al 1996). Это может говорить о том, что имеются и другие гены, обеспечивающие метилирование.
Механизм инактивации гена посредством ДНК метилирования только начинает проясняться. В настоящее время принята гипотеза, что белок MeCp2 (methylated-DNA binding protein 2) связывается с метилированной ДНК и включается в белковый комплекс, состоящий из гистоновых белков, деацетилазы и др. Этот белковый комплекс, в свою очередь, инициирует компактизацию хроматина, что не дает связаться факторам тракскрипции с промоторной областью и, следовательно, происходит инактивация гена (Jones et al. 1998,).
ДНК метилирование – физиологический процесс, который контролирует эпигенетическое наследование и экспрессию генов. Но этот процесс может быть нарушен и аберрантное метилирование CpG-островков является одной из причин, лежащих в основе изменений, наблюдаемых при старении и образовании раковых клеток.
Первые два гена, для которых было показано аберрантное метилирование, были кальцитонин и MyoD (Jones 1990). Функциональное значение этого явления ранее было неясно, поскольку оба гена не экспрессируются в большинстве тканей. Открытие привело к гипотезе, что метилирование промотора может быть одним из механизмов инактивации генов-супрессоров опухолевого роста в раковых клетках. Предположение было вскоре подтверждено обнаружением метилирования промоторной области известных генов-супрессоров опухолевого роста типа RB1, VHL и p16 в спорадических опухолях. В настоящее время список генов, инактивирующихся метилированием промоторной области, пополняется. Так было показано аномальное биаллельное метилирование промотора гена Н19
у пациентов с опухолью Вильмса. Также показано метилирование промоторной района р16 во многих типах опухолей (Myohanen et al. 1998, Huschtscha et al. 1998, Yao et al. 1998, Gonzalo et al. 1998, Esteller et al. 1999). В гепатокарциномах и аденокарциномах поджелудочной железы инактивация гена таким способом происходит почти в 70% случаев (Wilents et al 1998). Гиперметилирование промоторной области рецептора эстрогена (ER) обнаруживается в опухолях толстого кишечника (Issa et al. 1994). Транскрипционный фактор (MYOD), специфичный для мышечной ткани, гиперметилируется не только при раке мочевого пузыря, но и при раке толстого кишечника. Недавно клонированный ген N33, подвергается метилированию в том же типе опухолей, как и два других гена (ER и MYOD), более чем в 90% случаев (Ahuja et al. 1998). Тканевой ингибитор металлопротеиназы-3 (TIM-3) может подавлять рост опухоли, ангиогенез, прорастание и метастазирование (Cambers 1997). Показано, что аберрантное метилирование промоторного района этого гена происходит в различных опухолях: при немелкоклеточном раке легкого в 19%, раке молочной железы в 27%, раке толстой кишки в 27%, карциноме почки в 78% и глиобластоме в 26% случаев (Bachman 1999). В спорадических опухолях молочной железы обнаруживают инактивацию посредством метилирования генов BRCA1, MYOD и ER (Dobrovic et al. 1997, Ahuja 1998, Lapidus 1998). Таким образом, метилирование промоторной области может являться новым механизмом инактивации генов-супрессоров опухолевого роста.Роль метилирования ДНК в генезе опухолей была изучена на примере гена MНL1, который ответственен за развитие рака желудочно-кишечного тракта. В клетках опухоли выявляется большое количество генетических изменений - микросателлитные маркеры представлены одновременно многими разными аллелями
, дополнительными к двум родительским аллелям. Это явление получило название “нестабильности микросателлитов” (MSI) (Tibodeau et al., 1993) или “репликационных ошибок” (Aaltonen et al., 1993). Этот ген имеет гиперметилированный промоторный район. Обработка клеток опухоли метилтрансферазой, ингибирующей 5-аза-2’-деоксицитидин (5-azad-2’-C) приводила к деметилированию промотора MHL1, выработке MHL1 белка и восстановлению активности гена, вследствие чего опухоль претерпевала обратное развитие. Таким образом, было установлено, что гиперметилирование CpG-островков является ведущим в механизме инактивации MHL1.Точное или даже приблизительное количество генов, чья промоторная область гиперметилирована в опухолевых клетках, неизвестно. Трудности количественной оценки в том, что метилирование в клеточных линиях отличается от метилирования в нормальных тканях и опухолевых клетках организма, а также метилированию в различных опухолевых клетках подвергаются разные гены. В опухолях наиболее надежным методом для оценки частоты метилирования CpG-островков в настоящее время считается метод RLGS (restriction landmark genomic scanning). Используя этот метод, было показано, что в раковых клетках метилировано примерно 1-2% CpG-островков. Эти результаты, скорее всего, занижены, т. к. образцы опухолевой ткани очень часто бывают загрязнены нормальной (неопухолевой) тканью. Сastello et al. (1999) оценили частоту метилирования CpG-островков на 10 случайно отобранных генах в нескольких клеточных линиях. Частота метилирования в этих клетках составила 10-40%. Используя другие методы исследования, частота метилирования на этих же образцах, была оценена как 40-50%. Но независимо от используемого метода становится ясно, что в раковых клетках несколько тысяч CpG-островков метилировано. Остается только выяснить, сколько из них связаны с подавлением транскрипции гена, и какие из них вносят вклад в жизнедеятельность клетки.
Основными методами, применяемыми для определения метилирования CpG-островков, являются: 1) использование метилчувствительных рестриктаз (HpaII, HhaI, NotI, SacII, EagI, BssHII и др) с последующей амплификацией CpG-островка и, 2) бисульфитная модификация ДНК с последующей метил-специфической амплификацией или секвенированием. Метод основан на том, что бисульфит натрия преобразовывает все неметилированные цитозины в урацил, в то время как метилированные цитозины, стоящие перед гуанином остаются в немодифицированном состоянии.
![]()
![]()
![]()